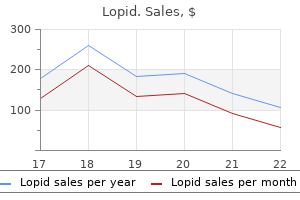
Clinical Director, University of Oklahoma School of Community Medicine
A massive Vd may point out that the drug is concentrated intracellularly medicine zetia cheap lopid 300 mg otc, with a ensuing low focus in the plasma symptoms 32 weeks pregnant 300 mg lopid purchase. After a weak base diffuses into a cell chi infra treatment lopid 300 mg for sale, a larger fraction is ionized in the more acidic intracellular fluid symptoms 0f colon cancer discount lopid 300 mg line. A massive Vd may outcome from sequestration into fat tissue, similar to occurs with the antimalarial agent chloroquine. Renal Clearance Renal clearance could be calculated as the renal excretion price divided by the plasma drug focus (see Box 2-2). Hepatic Clearance Hepatic clearance is tougher to determine than renal clearance. This is as a outcome of hepatic drug elimination consists of Drug Clearance Clearance (Cl) is the most fundamental expression of drug elimination. It is outlined as the quantity of physique fluid (blood) from which a drug is eliminated per unit of time. Whereas the clearance of a particular drug is constant, it is necessary to Chapter2 y Pharmacokinetics 21 the biotransformation and biliary excretion of mother or father compounds. For this purpose, hepatic clearance is normally decided by multiplying hepatic blood move by the arteriovenous drug focus distinction. First-order kinetics Plasma drug concentration (mg/L) eight Plasma drug focus (mg/L) Most drugs exhibit first-order kinetics, during which the rate of drug elimination (amount of drug eliminated per unit time) is proportional to the plasma drug concentration and follows an exponential decay operate. For drugs that exhibit first-order kinetics, the plasma drug concentration could be determined from the dose of a drug and its clearance. Disease, age, and other physiologic variables can alter drug clearance or quantity of distribution and thereby change the elimination half-life (see Chapter 4). The following principles pertain to zero-order kinetics: the rate of drug elimination is fixed. In many circumstances, the rationale that the rate of drug elimination is constant is that the elimination course of becomes saturated. In first-order kinetics (A), the rate of drug elimination is proportional to the plasma drug concentration. The kinetic order of a drug is derived from the exponent n within the following expression: [Drug]/t = -k e [Drug]n Zero-Order Kinetics the place represents change, [Drug] represents the plasma drug concentration, and t is time. When the drug is first administered, the speed of administration is way larger than the speed of elimination, because the plasma concentration is so low. As the drug continues to be administered, the rate of drug elimination 22 SectionI y PrinciplesofPharmacology Ct Ct C0 ke e = = = = = C0. Because the time to attain the regular state is dependent on the time it takes for the speed of drug elimination to equal to the speed of drug administration, the time to reach the steady state is a perform of the elimination half-life of the drug. Any first-order course of requires about 5 half-lives to be accomplished; thus the time to reach the steady-state drug focus is about 5 drug half-lives. If the half-life of a drug adjustments, then the time required to attain the steady state also changes. Note that the time required to reach the steady state is unbiased both of the drug dose and the rate or frequency of drug administration. A drug administered intermittently will accumulate to a gentle state at the identical rate as a drug given by continuous infusion, however the plasma drug concentration will fluctuate as every dose is absorbed and eliminated. The average steadystate plasma drug concentration with intermittent intravenous administration would be the identical as if the equal dose were administered by steady infusion. With intermittent oral administration, the bioavailability of the drug may even affect the steady-state plasma focus. The elimination half-life (t1/2) is the time required to scale back the plasma drug focus (C) by 50%. Loading Dose A loading dose, or priming dose, is given to rapidly establish a therapeutic plasma drug concentration. The loading dose may be calculated by multiplying the amount of distribution by the desired plasma drug concentration. The loading dose, which is larger than the maintenance dose, is mostly administered as a single dose, but it could be divided into fractions which might be given over a number of hours. A divided loading dose is usually used for medicine that are more toxic, for example, digitalis glycosides used to deal with congestive heart failure. Maintenance Dose A maintenance dose is given to set up or keep the desired steady-state plasma drug concentration. For medication given intermittently, the upkeep dose is certainly one of a sequence of doses administered at regular intervals. The quantity of drug to be given is based on the precept that on the regular state the speed of drug administration equals the rate of drug elimination. The half-life is often decided from the plasma drug focus curve proven here. The clearance (Cl) is the quantity of fluid from which a drug is eliminated per unit of time. Eventually, as the plasma focus rises sufficiently, the rate of drug elimination equals the rate of drug administration. The upkeep dose is then calculated as the rate of drug elimination multiplied by the dosage intervals. If the drug is administered orally, its bioavailability should even be included in the equation. It is calculated by dividing the drug dose by the plasma drug focus at time zero. The renal clearance of a drug can be calculated by dividing the renal excretion rate by the plasma drug concentration. If drug elimination mechanisms (biotransformation and excretion) turn out to be saturated, a drug can exhibit zero-order kinetics, in which the rate of drug elimination is fixed. The half-life is the time required for the plasma drug focus to lower by 50%. The clearance is the volume of plasma from which a drug is eliminated per unit of time. Factors that cut back bioavailability include incomplete pill disintegration and first-pass and gastric inactivation of a drug. It takes about 4 to 5 drug half-lives to achieve the steady-state situation. A, the steady-state plasma drug concentration is pro- portional to the dose administered per unit of time. B, the steady-state plasma drug concentration is immediately proportional to the half-life (and is inversely associated to clearance). With intermittent drug administration, nevertheless, the plasma concentrations fluctuate between doses, and the dimensions of fluctuations increases because the dosage interval will increase. D, Plasma drug concentrations after intermittent oral administration are affected by the charges of drug absorption, distribution, and elimination. If only one dose is given, the height in plasma drug focus is adopted by a steady decline in the curve. Chapter2 y Pharmacokinetics 25 � A loading dose is a single or divided dose given to rapidly set up a therapeutic plasma drug focus. The dose can be calculated by multiplying the quantity of distribution by the desired plasma drug focus. The quantity of distribution of a drug shall be higher if the drug (A) is more ionized inside cells than in plasma (B) is run very quickly (C) is extremely ionized in plasma (D) has poor lipid solubility (E) has a excessive molecular weight evaluation Questions 1. If food decreases the speed but not the extent of the absorption of a specific drug from the gastrointestinal tract, then taking the drug with meals will lead to a smaller (A) space beneath the plasma drug focus time curve (B) maximal plasma drug focus (C) time at which the maximal plasma drug focus occurs (D) fractional bioavailability (E) whole clearance 2. If a drug displays first-order elimination, then (A) the elimination half-life is proportional to the plasma drug concentration (B) the drug is eradicated at a relentless fee (C) hepatic drug metabolizing enzymes are saturated (D) drug clearance will increase if the plasma drug concentration increases (E) the speed of drug elimination (mg/min) is proportional to the plasma drug concentration three. After an individual ingests an overdose of an opioid analgesic, the plasma drug concentration is discovered to be 32 mg/L. Moreover, the time at which the maximal plasma drug concentration occurs will enhance. The reply is E: the speed of drug elimination (mg/min) is proportional to the plasma drug focus. The dose required to establish a goal plasma drug concentration is calculated by multiplying the clearance by the target focus and dosage interval. When a drug is extra ionized inside cells, the drug becomes sequestered within the cells and the amount of distribution can turn into quite massive.
The ectropion can generally be a reason for post-coital bleeding or mucoid vaginal discharge medications januvia lopid 300 mg purchase with visa, and mostly happens throughout being pregnant or within the woman taking the combined oral contraceptive tablets symptoms before period buy lopid 300 mg overnight delivery. Since the transformation zone is the commonest website for cervical carcinoma inoar hair treatment generic 300 mg lopid overnight delivery, it is necessary to stroke treatment 60 minutes lopid 300 mg generic with amex brush this website adequately when performing a cervical smear. The endocervical glands are liable for producing mucus, whose consistency varies throughout numerous phases of the menstrual cycle. At ovulation, the consistency of the mucus adjustments and it becomes extra thin and stretchable so that sperms can penetrate through it and fertilisation can happen. The cervix is mostly composed of fibrous tissue, which mainly comprises of collagen and a small amount of elastin. Posteriorly the peritoneum passes downwards from the uterine body to cover the posterior floor of the supravaginal cervix and the upper third of the posterior vaginal wall. The peritoneum is then mirrored on to the rectum, ensuing in the formation of the rectouterine pouch or the pouch of Douglas. Lymphatic drainage of the cervix: the lymphatics from the cervix move either laterally in the base of the broad ligament or posteriorly alongside the uterosacral ligaments to attain the sidewall of the pelvis. Most of the vessels drain to the interior iliac, obturator and exterior iliac nodes, but some vessels also move directly to the widespread iliac and lower para-aortic nodes. Innervation of the cervix: Pain from the cervix is carried by pelvic splanchnic nerves, hence there could be bradycardia during cervical dilatation. The primary blood supply to the uterus is through the uterine artery (one on every side), which is a department of the anterior trunk of the inner iliac artery. The uterine artery first runs downwards on the lateral wall of the pelvis, in the same direction because the ureter; then turns inwards and forwards, lying in the base of the broad ligament. By taking such a course, the uterine artery crosses above the ureter at a distance of about 2 cm from the uterus, on the stage of the interior os. On reaching the wall of the uterus, the uterine artery turns upwards to run tortuously alongside the lateral uterine sidewalls to reach the higher a part of the uterus close to the doorway of fallopian tubes. It continues to move alongside the decrease border of the fallopian tube where it ends by anastomosing with the ovarian artery, a direct department of the belly aorta. In this a half of its course, the uterine artery offers rise to branches, which run transversely and cross into the myometrium. Blood provide to anterior and posterior partitions is offered by the arcuate arteries, which run circumferentially across the uterus. From the arcuate vessels, branches often identified as the radial arteries arise at proper angles. From the basal arteries, spiral and straight arterioles of the endometrium are derived. The arcuate artery to the cervix is also referred to as the round artery of the cervix. The uterine artery offers off a small descending department that supplies the cervix and the vagina. The uterine artery also provides branches to the fallopian tube and ureter as it crosses it. Cervicovaginal branches anastomose with vaginal arteries to kind the azygos arteries of the vagina. Trophoblastic invasion of the spiral vessels throughout second trimester in regular being pregnant is liable for a 10-fold improve in blood move. Due to this trophoblastic invasion, the small muscular spiral arteries get transformed into massive vascular channels, which transform the uteroplacental circulation right into a low-resistance system. The uterine vein follows the uterine artery all along its course in the broad ligament and varieties a uterine venous plexus on all sides of the cervix. The uterine venous plexus is related with the superior rectal vein, thereby forming a portal systemic anastomosis. Parasympathetic fibres of the uterus are derived from the pelvic splanchnic nerves (S2�S4). The afferent fibres mainly ascend via the inferior hypogastric plexus to enter the spinal cord by way of T10�T12 and L1 nerve fibres. The uterus contains alpha-receptors, which cause contractions in the pregnant uterus, and betareceptors, which cause leisure within the non-pregnant uterus. Thus the uterine contraction and leisure is under the control of sympathetic nervous system. Hypogastric plexus is a plexus of nerves, which provides the viscera of the pelvic cavity. It contributes branches to the uterine/vaginal plexus in females, vesical plexus in each males and females and to the prostatic plexus within the males. The inferior hypogastric plexus is a paired structure, which lies between the pelvic viscera (vagina and rectum) and the pelvic wall in females and on the either aspect of the rectum in males. It extends into the base of the broad ligament of the uterus and lies between the two iliac vessels. The sources of the inferior hypogastric plexus are as following: T Hypogastric nerve, which is a continuation of the superior hypogastric plexus on the either side T Sacral splanchnic nerves (postganglionic sympathetic axons) T Pelvic splanchnic nerves (preganglionic parasympathetic axons from the ventral main rami of spinal nerves S2�S4). The testes are responsible for producing sperms and male hormones (mainly testosterone) that regulate reproductive organ development. The seminiferous tubules of testis produce spermatozoa, which acquire their mobility in the epididymis. Primary spermatocytes are fashioned from spermatogonium, which divide into two secondary spermatocytes, which ultimately divide into four spermatids. In the grownup testis, Leydig cells express aromatase (P450arom) and actively synthesise oestradiol. Lymphatic drainage: the testicular lymphatics cross together with the arteries to the para-aortic group of lymph nodes in the region of the renal arteries. Lymph from the vulva (in females), scrotum (in males) and decrease limbs (in both) passes to the superficial after which on to the deep inguinal nodes. Pelvic Organs: Part of the Gastrointestinal Tract Rectum the rectum extends from the extent of the body of the third sacral vertebra to the anorectal line. The rectum varies from 10 cm to 15 cm in length, while the circumference varies from 15 cm at the rectosigmoid junction, to 35 cm or more at its widest ampullary portion. The decrease end of the rectum lies somewhat beneath and in front of the tip of the coccyx and turns into continuous with the anal canal. The decrease part of the rectum, which is wider than the upper half, is called the ampulla. Lymphatic Drainage the fundus and higher a part of the uterine body drain into the lumbar (aortic) nodes, the lower part of the physique into the external iliac, nodes, and the cervix into the external and inside iliac and the sacral nodes. The lymphatic drainage of the fundus of the uterus is into the para-aortic nodes (at the extent of first lumbar vertebra). Nearly all the lymphatic vessels from the corpus uteri be part of these leaving the cervix and due to this fact attain similar teams of nodes. Male Internal Genitalia Testes Each testis (one on either side) is an oval-shaped construction about 4 cm in its longest (vertical) diameter and lies in the scrotal sac of each facet respectively. The outer surface of the testis is fashioned by a dense fibrous membrane known as the tunica albuginea. The substance of testis comprises of a lot of lobules, which are separated from each other by septa. These tubules are lined by an epithelium, the cells of which are concerned with the manufacturing of spermatozoa. The upper a part of the rectum is directed backwards, and the lower half is directed forwards. While passing downwards from the midline, the rectum deviates first to the right, then to the left and again to the best, finally returning to the center line at its lower finish. There is a sudden change in path of the alimentary canal at the junction of the rectum with the anal canal. The anorectal junction lies on the level of the pelvic diaphragm, with the rectum mendacity above the pelvic diaphragm within the true pelvis, and the anal canal lying below it within the perineum. Folds in the Rectum the mucous membrane of the rectum shows numerous transverse folds. Peritoneal Relations the upper part of the rectum is covered with peritoneum in entrance and at the sides; the center half is covered in entrance only; the decrease part lies beneath the extent of the rectovaginal pouch and subsequently is devoid of peritoneal masking. Interior of the Anal Canal the upper 15 mm or so of the anal canal is lined by mucous membrane.
Discount 300 mg lopid amex. Oral Manifestations of HIV: Case Studies.
Receptor Regulation and Drug Tolerance Efficacy the ability of a drug to initiate a cellular impact known as intrinsic exercise or efficacy treatment nail fungus lopid 300 mg buy generic line. Drugs that have each receptor affinity and efficacy are called agonists treatment jerawat di palembang order lopid 300 mg online, whereas medicine which have receptor affinity but lack efficacy are referred to as antagonists medicine mound texas lopid 300 mg buy discount. With a few lessons of drugs medicine xalatan 300 mg lopid order otc, corresponding to agonists and antagonists on the -adrenoceptor, the precise molecular structures liable for affinity and efficacy are identified. Both agonists and antagonists have common components enough for receptor affinity, however solely agonists have the construction required for efficacy. Full agonists can produce the maximal response obtainable in a tissue and due to this fact have maximal efficacy. Inverse agonists, that are also referred to as unfavorable antagonists, are concerned in a particular sort of drug-receptor interplay. The effect of inverse agonists relies on the finding, in some circumstances, that Receptors can bear dynamic changes with respect to their density (number per cell) and their affinity for medicine and different ligands. Phosphorylation of the receptor reduces the G protein�coupling efficiency and alters the binding affinity. This short-term effect of agonist publicity is recognized as desensitization or tachyphylaxis. Through internalization and regulation of the receptor gene, the variety of receptors on the cell membrane decreases. In distinction, continuous or repeated exposure to antagonists initially can enhance the response of the receptor, referred to as supersensitivity. With chronic exposure to antagonists, the variety of receptors on the membrane floor (density) increases by way of up-regulation. Drug tolerance is seen when the same dose of drug given repeatedly loses its impact or when greater doses are wanted to obtain a beforehand obtained impact. Receptor downregulation is usually responsible for pharmacodynamic tolerance, which describes variations to persistent drug publicity at the tissue and receptor degree. Pharmacodynamic tolerance is distinct from pharmacokinetic tolerance in that the latter is caused by accelerated drug elimination, usually ensuing from an up-regulation of the enzymes that metabolize the drug. Disease states can alter the number and performance of receptors and thereby affect the response to drugs. For example, myasthenia gravis is an autoimmune dysfunction in which antibodies destroy the nicotinic receptors in skeletal muscle, leading to impaired neurotransmission and muscle weak spot. This condition is handled by administration of nicotinic receptor agonists (see Chapter 6). The relationship between the focus of a drug at the receptor website and the magnitude of the response is identified as the dose-response relationship. Depending on the aim of the studies, this relationship could be described in phrases of a graded (continuous) response or a quantal (allor-none) response. The dose-response Z curves for a therapeutic effect (sleep) and a poisonous effect (death) of a drug are compared. Drug this a partial agonist and subsequently is incapable of producing the same magnitude of effect as a full agonist. X, Agonist alone; Y, agonist in the presence of a aggressive antagonist; Z, agonist within the presence of a noncompetitive antagonist. In graded dose-response relationships, the response elicited with each dose of a drug is described in terms of a percentage of the maximal response and is plotted towards the log dose of the drug. Graded dose-response curves illustrate the relationship amongst drug dose, receptor occupancy, and the magnitude of the ensuing physiologic impact. For a given drug, the maximal response is produced when all the receptors are occupied, and the half-maximal response is produced when 50% of the receptors are occupied. In some cases, fewer than 50% of complete receptors will be occupied but still give the half-maximal response. This is because only a fraction of the total receptors are wanted to produce the maximal response. Potency is a attribute of drug action helpful for comparing totally different pharmacologic agents. Potency is basically determined by the affinity of a drug for its receptor, as a outcome of drugs with greater affinity require a lower dose to occupy 50% of the useful receptors (or much less if spare receptors are present). A full agonist has maximal efficacy, whereas a Graded Dose-Response Relationships partial agonist has lower than maximal efficacy and is incapable of manufacturing the same magnitude of impact as a full agonist, even at the very highest doses. When a partial agonist is administered with an agonist, the partial agonist could act as an antagonist by stopping the agonist from binding to the receptor and thereby reducing its effect. An antagonist, by definition, has no efficacy in this sense however could be an efficient medication, as in the utilization of a -adrenoceptor antagonist (-blocker) to deal with hypertension. The effect that an antagonist has on the dose-response curve of an agonist is determined by whether or not the antagonist is competitive or noncompetitive. A competitive antagonist binds reversibly to a receptor, and its effects are surmountable if the dose of the agonist is elevated sufficiently. Quantal Dose-Response Relationship In quantal dose-response relationships, the response elicited with each dose of a drug is described when it comes to the cumulative proportion of topics exhibiting an outlined allor-none impact and is plotted in opposition to the log dose of the drug. An instance of an all-or-none effect is sleep or not-asleep when a sedative-hypnotic agent is given. The regulation of mass motion explains the relationship between (A) dose of drug and physiologic response (B) the focus of drug and the affiliation or dissociation of drug-receptor complicated (C) receptors and the rate of signal transduction (D) an enzyme and ligands that inhibit the enzyme (E) graded and quantal dose-response curves 4. In a log dose-response plot, drug efficacy is decided by the maximal peak of the measured response on the effect axis, whereas drug efficiency is determined by (A) variety of animals exhibiting an all-or-none response (B) signal transduction pathway (C) method, including the affinity of the drug and the variety of drug receptors (D) position of the curve along the log-dose axis (E) steepness of the dose-response curve 5. Agonists have each affinity and efficacy, whereas antagonists only have receptor affinity. The ratio of the median lethal dose (toxic dose) to the median efficient dose (therapeutic dose) known as the therapeutic index, which is an indication of the margin of safety of a drug. Pharmacodynamics is the study of the detailed molecular pathway starting from the drug (ligand) binding to its receptor, the activation of effector molecules. The reply is B: the concentration of drug and the association or dissociation of drug-receptor complex. Potency and efficacy could be decided from a graph of the log dose-response curve by visual inspection. The placement of the curve alongside the log-dose axis or x-axis determines potency such that curves to the left characterize more potent medicine than curves to the best. Whereas some agents are potent and efficacious, these two traits of drug action review Questions 1. The description of molecular events initiated with the ligand binding and ending with a physiologic effect is identified as (A) receptor down-regulation (B) signal transduction pathway (C) ligand-receptor binding (D) regulation of mass action (E) intrinsic activity or efficacy 2. An agonist acts at its receptor to activate the signal transduction pathway and produce an effect. A partial agonist binds to the receptor and activates the signal transduction pathway, however not to the maximal diploma. Federal regulations require that extensive toxicity studies in animals be carried out to predict the risks that shall be associated with administering the drug to healthy human subjects and sufferers. The value of the preclinical research relies on the proven correlation between drug toxicity in animals and humans. As outlined in Table 4-1, the studies involve short-term and long-term administration of the drug and are designed to decide the danger of acute, subacute, and chronic toxicity, as properly as the risk of terato genesis, mutagenesis, and carcinogenesis. After animals are treated with the new drug, their conduct is assessed and their blood samples are analyzed for indications of tissue damage, metabolic abnormalities, and immunologic results. Studies in animals might not reveal all the opposed effects that will be found in human topics, either due to the low incidence of explicit effects or due to variations in susceptibility amongst species. This implies that some adverse reactions is most likely not detected until the drug is run to humans. The overwhelming success in fashionable pharmacotherapy in treating illness states attests to the protection and efficacy of prescribed brokers. However, drugs may also be poisons inflicting unwanted antagonistic results, and drugs can kill. This chapter begins with a description of drug development and the processes for evaluating drug safety and efficacy after which discusses the various types of opposed effects and interactions which may be caused by medication. Considerations for specific populations, such because the neonate and the elderly, are highlighted, and the legal guidelines regarding drug use and abuse are briefly reviewed. Discovery and Characterization New drug compounds are synthesized de novo or are isolated from a pure product, or a mixture of the 2 as in semisynthetic compounds. Synthetic medication may be patterned after other medication with known pharmacologic activity, or their construction could additionally be designed to bind a specific receptor and primarily based on pc modeling of the drug and receptor.
The adverse results of medication could additionally be caused by extreme pharmacologic effects medicine kidney stones generic 300 mg lopid visa, hypersensitivity reactions medicine review trusted 300 mg lopid, or other mechanisms answerable for organ toxicities treatment keloid scars lopid 300 mg buy on line. Drug interactions happen when one drug alters the pharmacologic properties of another drug medications japan travel lopid 300 mg order amex. Most interactions are brought on by pharmacokinetic results, significantly inhibition or induction of drug biotransformation. Age, disease, being pregnant, and lactation are elements that must be thought-about in drug selection and dosage. The very younger and the very old tend to have an increased sensitivity to therapeutic brokers, normally due to a decreased capacity to get rid of drugs. An advertisement in an area newspaper seeks to enroll 20 sufferers with arthritis in a medical examine that would be the primary time that a new drug can be tested in individuals with this illness. Which of the next drug interplay mechanisms is most probably to result in sustained elevations of plasma drug concentrations and drug toxicity Elderly individuals might have altered drug disposition due to (A) markedly decreased absorption of many medicine (B) higher volumes of distribution for water-soluble drugs (C) accelerated renal excretion of ionized medicine (D) elevated permeability of the blood-brain barrier (E) reduced capability to oxidize medicine Answers And explAnAtions 1. Phase I studies are done to set up security and pharmacokinetics in wholesome subjects, often students within the well being professions. These medicine should still be abused by diversion, the act of illegally acquiring prescribed drugs by sale or theft. Teratogenic medication could trigger fetal malformations if taken by a pregnant woman during this interval. These malformations embody cleft palate, malformation of fingers and toes, coronary heart defects, facial abnormalities, and skeletal deformities. Drug-induced labor or jaundice is primarily of concern over the last trimester of pregnancy. Inhibition of drug-metabolizing enzymes will increase the half-life and plasma concentrations of affected drugs, thereby posing a danger of toxicity. Displacement of a drug from plasma proteins or inhibition of P-glycoprotein may enhance plasma levels briefly until the speed of elimination will increase. Conjugative metabolism is comparatively unchanged in the aged, but oxidative drug metabolism is often lowered. The elderly are most likely to have the next percentage of physique fat than youthful adults and due to this fact have increased volumes of distribution of fat-soluble drugs. The sympathetic nervous system tends to discharge as a unit, producing a diffuse activation of target organs. Preganglionic, sympathetic neurons synapse with a giant quantity of postganglionic neurons, which contributes to widespread activation of the organs during sympathetic stimulation. In addition, the discharge of epinephrine and norepinephrine from the adrenal medulla into the circula tion allows the activation of target tissues throughout the body, together with some tissues in a roundabout way innervated by sympathetic nerves. In distinction, the parasympathetic system can discretely activate particular target tissues. This is partly due to the low ratio of postganglionic fibers to preganglionic fibers in the para sympathetic system. Activation of the sympathetic system produces the "struggle or flight" response in response to threatening situa tions. In this reaction, cardiovascular stimulation supplies muscular tissues with oxygen and fuels required to assist vigorous bodily exercise, while activation of glycogenolysis and lipolysis releases the necessary vitality substrates. The para sympathetic system is typically referred to as the "rest and digest" system, because it slows the guts rate and promotes more vegetative capabilities, such as digestion, defecation, and micturition. Many parasympathetic results (including pupillary constriction, bronchoconstriction, and stimulation of intestine and bladder motility) are attributable to smooth muscle contraction. The central nervous system consists of the mind and spinal twine, whereas the peripheral nervous system consists of the autonomic nervous system and the somatic nervous system. Drugs alter nervous system perform primarily by have an result on ing neurotransmitters or their receptors. In some cases medication alter the synthesis, storage, release, inactivation, or neuronal reuptake of neurotransmitters. Most medication are relatively selective for a selected neurotransmitter or recep tor. The results produced by a drug depend partly on the distribution of the affected neurotransmitters in the central and peripheral nervous techniques. The actions of some medicine are localized to both the central or the peripheral nervous system, but different drugs. This chapter reviews the anatomy and physiology of the peripheral nervous system and introduces the mechanisms by which medication affect nervous system perform. Both the autonomic and the somatic nervous techniques are managed by the central nervous system. The autonomic nervous system is regulated by mind stem facilities liable for cardiovascular, respir atory, and different visceral features. The somatic nervous system is activated by corticospinal tracts, which originate in the cerebral motor cortex, and by spinal reflexes. Somatic Nervous System Autonomic Nervous System the somatic nervous system consists of the motor neurons to the skeletal muscle. These neurons have a single nerve fiber that releases acetylcholine on the neuromuscular junction. The autonomic nervous system consists of sympathetic and parasympathetic divisions. In the sympathetic nervous system, nerves come up from the thoracic and lumbar spinal twine and have a brief preganglionic fiber and a protracted publish ganglionic fiber. Most of the ganglia are located within the para vertebral chain adjoining to the spinal wire, however a number of prevertebral ganglia (the celiac, splanchnic, and mesenteric ganglia) are positioned extra distally to the spinal twine. The parasympathetic nerves have lengthy preganglionic fibers and short postganglionic fibers, with the ganglia usually situated in the innervated organs. The parasympathetic nervous system consists of cranial and sacral nerves with long preganglionic and quick postganglionic fibers. The sympathetic nervous system consists of thoracic and lumbar nerves with short preganglionic and lengthy postganglionic fibers. The sympathetic system contains the adrenal medulla, which releases norepinephrine and epinephrine into the blood. The terms adrenergic and cholinergic discuss with neurons that launch norepinephrine or acetylcholine, respectively. Acetylcholine is the transmitter at all autonomic ganglia, at parasympathetic neuroeffector junctions, and at somatic neuromuscular junctions. It is also the transmitter at a few sympathetic neuroeffector junctions, including the junctions of nerves in sweat glands and vasodilator fibers in skeletal muscle. The presence of acetylcholine in several types of autonomic and somatic synapses contributes to the dearth of specificity of medication performing on acetylcholine neurotransmission. Although norepinephrine (noradrenaline) is the first neurotransmitter at most sympathetic postganglionic neu roeffector junctions, epinephrine (adrenaline) is the princi pal catecholamine launched from the adrenal medulla in response to activation of the sympathetic nervous system. The transmitters released by these neurons embrace neuropeptide Y, vasoactive intestinal polypeptide, enkephalin, substance P, serotonin (5hydroxytryptamine), adenosine triphosphate, and nitric oxide. In some tissues, adenosine triphosphate launched by these neurons is converted to adenosine, which might then activate adenosine receptors in a quantity of tissues (see Chapter 27). The acetylcholine receptors have been divided into two types, primarily based on their selective activation by considered one of two plant alkaloids. Muscarinic (M) receptors, which are acetylcho line receptors activated by muscarine, are primarily located at parasympathetic neuroeffector junctions. They are present in all autonomic ganglia, at somatic neuro muscular junctions, and within the mind. Muscarinic receptors are subdivided based on molecular and pharmacologic crite ria. Activation of the M3 receptor produces smooth muscle contraction (except sphincters) and gland secretion. The M1 receptor is primarily concerned with modulation of neuro transmission at central and peripheral sites. Activation of nicotinic receptors in autonomic ganglia excites neurotrans mission, whereas activation of these receptors in skeletal muscle causes muscle contraction. Sympathetic effects are mediated by adrenoceptors (), adrenoceptors (), or muscarinic receptors (M). The two kinds of adrenoceptors, known as adrenoceptors and -adrenoceptors, could be activated or blocked by medicine known as adrenoceptor agonists and adrenoceptor antagonists, respectively.